- Published on
Embeddings to Phylogenies
- Authors

- Name
- RostLab
- @rostlab

- Name
- Dr. Ivan Koludarov
Our lab, endowed with extensive experience in harnessing artificial intelligence for understanding protein structures, recently conducted an investigation into the hidden correlation between proteins and phylogenetics - the study of evolutionary relationships. Understanding phylogenetic relationships is not only instrumental to comprehend the evolution of life on Earth, but also holds significant implications for drug development by elucidating natural changes and their effects. However, teasing apart these relationships from protein sequences is a complex and demanding task due to the immense diversity and intricate structures inherent to proteins.
Our study revolved around one of our previously compiled datasets for a vertebrate kallikrein gene family, and more specifically, the esm-2 embeddings derived from it. We expanded our investigation by comparing these to esm-2 embeddings generated from kinase and phosphatase datasets.
Our initial step was to confirm the relationship between the information within these embeddings and sequence similarity. We randomly selected 1000 samples from Uniclust30 and Uniclust90 datasets, both of which cluster proteins based on sequence similarity. By comparing the embeddings with Needleman Wunsch sequence similarity scores, we confirmed that our embeddings did indeed contain relevant sequence similarity information. To ensure robustness, we evaluated the correlation of sequence similarity with euclidean, cosine, and chebyshev distances, employing Pearson’s, Kendall, and Spearman’s correlation coefficients.
Continuing our investigation, we took each dimension of our 1024-dimensional embedding vectors and tested them individually against sequence similarity scores using Spearman’s correlation, which had shown reliable performance in our previous analysis. We then expanded this analysis to include pairs of dimensions, ranging from two to nine dimensions. Linear regression analyses were conducted on the 1000 Uniclust90 embeddings to find the best fitting linear relationship between the similarity of the embeddings.
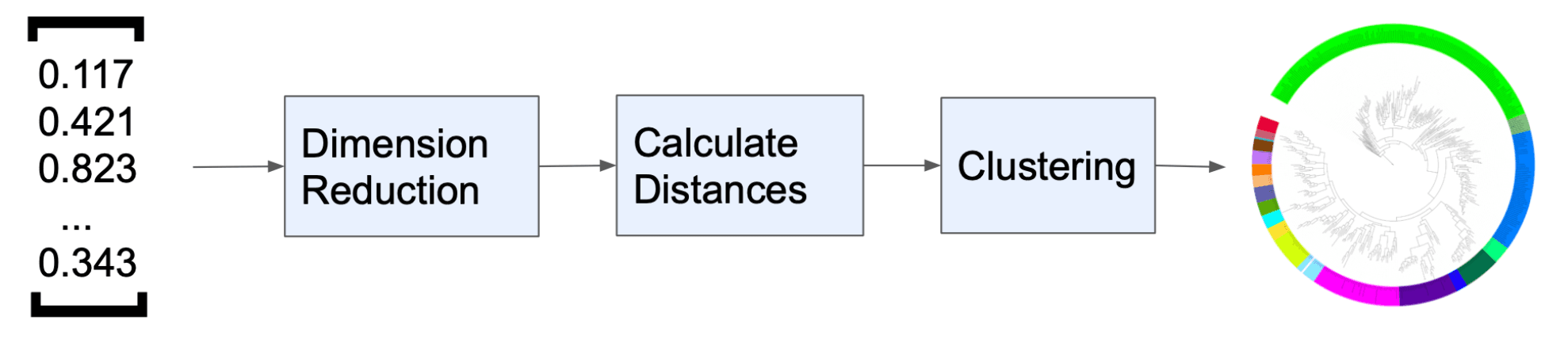
In an attempt to simplify our complex data, we looked at several dimension reduction techniques. We first attempted to implement a Variational Autoencoder (VAE), inspired by previous work, to reduce the dimensions and eliminate noise in the embeddings. However, the VAE model, despite various configurations and activation functions, did not yield the anticipated improvement in our data. We then turned to traditional dimension reduction techniques, namely t-SNE and PCA, for comparison.
Following the dimension reduction process, e generated a distance matrix using cosine, euclidean, manhattan, and TS-SS metrics on the dimension-reduced latent space. Then, using this distance matrix, we constructed phylogenetic trees using both neighbor joining (NJ) and UPGMA methods.
Finally, we compared our generated trees with the reference tree created with ExaBayes after building the MSA on the KLK protein sequences. We evaluated our trees, including the tree constructed without any dimension reduction techniques (termed the non-trained tree), based on the robinson-foulds (RF) distance, euclidean distance, and symmetric difference, as well as visually inspecting the trees.
Through this investigation, we found that the initial protein embeddings, even without additional dimension reduction, provided substantial insight into the phylogenetic relationships of proteins, serving as a significant milestone in our ongoing quest to decipher the evolutionary narrative of life on Earth and improve drug development strategies.
We presented our results in a poster at ISMB/ECCB 2023 in Lyon (Schmucklermann et al., 2023).
References: